
| 2013-2 |
|
|
|
|
|
|
| 手法: ・平面波基底PAW法に基づく 第一原理分子動力学計算 (VASPコード) ・機械学習: 既知のデータから有用な 特徴を抽出し、新たなデータを予測する 技術。学習モデルとして サポートベクター回帰(SVR)を採用。 |
|
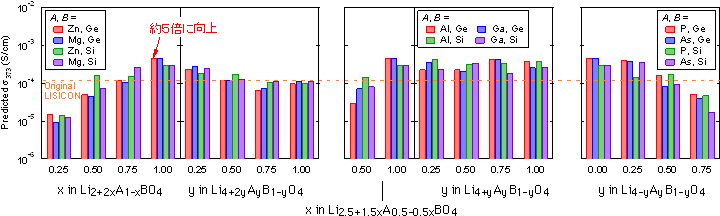 |
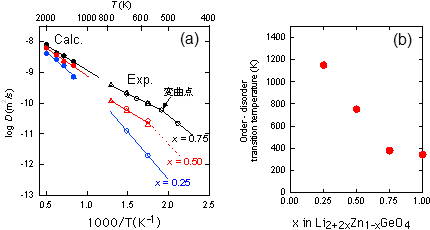
| 図2. 第一原理計算のデータと実験の伝導度データを入力情報とする機械学習によって 予測された 373 Kにおける γ-LISICON系固体電解質のイオン伝導度 |
|
|
|